Lithium ion batteries have aided the revolution in microelectronics and have become the choice of power source for portable electronic devices. Their triumph in the portable electronics market is due to the higher gravimetric and volumetric energy densities offered by them compared to other rechargeable systems. The higher energy density is due to the higher operating voltages of ∼4 V resulting from the use of water-free, nonaqueous electrolytes compared to the use of aqueous electrolytes in other systems that limit the operating voltages mostly to <2 V. Lithium ion batteries have also begun to enter the electric vehicle market and are being intensively pursued for grid energy storage as well. Energy, power, charge–discharge rate, cost, cycle life, safety, and environmental impact are some of the parameters that need to be considered in adopting lithium ion batteries for various applications. (1-8) While energy density is the most important factor for portable electronics, cost, cycle life, and safety also become critical parameters along with energy density (driving distance between charges) for electric vehicles. On the other hand, cost, cycle life, and safety become more important than energy density for grid-energy storage. It is desirable to have a fast charge–discharge rate for all three applications.
The performance parameters presented above are largely determined by the properties and characteristics of the component materials used in assembling the batteries as well as the cell engineering and system integration involved. The characteristics of the materials employed rely on the underlying chemistry associated with the materials. Presently, the commercial lithium ion technology is largely limited to cells with gravimetric energy densities of <250 W h kg–1 and volumetric energy densities of <650 W h L–1. While the energy densities are not critical for grid storage, volumetric energy densities are often more important for portable electronics and electric vehicles. There is immense interest around the world to push the energy densities to as high as ∼500 W h kg–1 and >1,000 W h L–1. Accomplishing this goal is challenging; it will need innovations both in the component materials used in the cell and in the engineering involved in fabricating the cells. It should be recognized that the incremental improvements made in energy density since the first announcement in 1991 by Sony Corporation of the commercialization of lithium ion technology is largely due to the progress in engineering as the component electrode materials still remain the same with minor modifications. The sections below provide the current status and where the technology is heading, followed by conclusions.
Current Lithium Ion Technology
Anodes
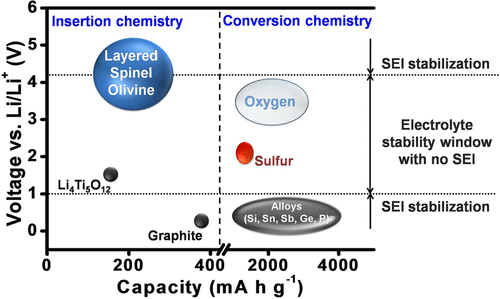
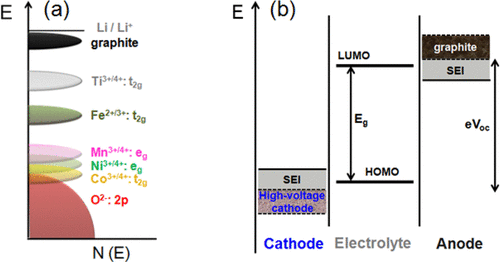
The current lithium ion technology is based on insertion-compound cathodes and anodes (Figure 1) and organic liquid electrolytes (e.g., LiPF6 salt dissolved in a mixture of organic solvents, such as ethylene carbonate (EC), dimethyl carbonate (DMC), diethyl carbonate (DEC), ethyl methyl carbonate (EMC), etc.). With an operating voltage close to that of Li/Li+ (∼0.1 V vs Li/Li+) and a capacity of 372 A h kg–1, corresponding to the insertion of one Li per six carbon atoms to give LiC6, graphite (Figure 2) has dominated as an anode in commercial lithium ion cells for the past 25 years. (9) Although the redox energy of graphite lies above the lowest unoccupied molecular orbital (LUMO) of the organic electrolytes used, the formation of a stable solid electrolyte interphase (SEI) layer on the graphite surface in reaction with the electrolyte solvents provides the stability for its operation with a long life (Figure 3). However, the slow lithium diffusion through the SEI could lead to lithium dendrite formation on the graphite surface and internal shorts resulting in catastrophic safety hazards as its operating voltage is close to that of Li/Li+, particularly under conditions of fast charge and at low temperatures. The redox energy of an alternative insertion-reaction anode Li4Ti5O12 with the spinel structure lies below the LUMO of the electrolyte (Figure 3), i.e., within the electrolyte stability window without the formation of an SEI (Figure 1). With no SEI and with a negligible volume change (<1%), Li4Ti5O12 offers long cycle life. Unfortunately, with an operating voltage of 1.5 V vs Li/Li+ and a limited capacity of ∼160 A h kg–1, (10) it reduces the cell energy density drastically. Nevertheless, it is being employed in cells for grid storage.
Figure 1
Figure 1. Capacity and voltage ranges of anode and cathode materials for lithium-based batteries. The voltage stability window for the currently used liquid electrolytes in lithium ion batteries and the possibility to widen the stability window by the formation of optimal SEI layers on the electrodes are indicated.
Figure 2
Figure 2. Crystal structures of graphite LixC6, layered LiMO2 (M = Mn, Co, and Ni), spinel LiMn2O4, and olivine LiFePO4.
Figure 3
Figure 3. (a) Positions of the various redox couples relative to the top of the oxygen:2p band and (b) schematic energy levels of an anode, cathode, and electrolyte in an open circuit. The possibility to widen the stability window by the formation of optimal SEI layers on the electrodes are indicated in panel b.
Cathodes
For the cathode, there are three choices: layered LiMO2 (M = Mn, Co, and Ni), (11) spinel LiMn2O4, (12) and olivine LiFePO4(13) (Figure 2). Each of these three cathodes have their advantages and disadvantages. The layered structure gives the highest practical capacity (currently up to ∼180 A h kg–1) among the three, but suffers from structural and/or chemical instabilities during cycling depending on the chemical composition and state of charge (lithium content in the electrode). The structural instability arises from a migration of the transition-metal ions from the octahedral sites of the transition-metal layer to the octahedral sites of the lithium layer via a neighboring tetrahedral site. (14) Mn3+ with a low octahedral-site stabilization energy (OSSE, i.e., a small difference between the crystal field stabilization energies in the octahedral and tetrahedral sites), for example, easily migrates and suffers from a structural transition from layered to spinel phase during cycling. Co3+ with a high OSSE offers excellent structural stability, but it suffers from poor chemical stability on extracting >50% lithium from LiCoO2 (>50% charge). The chemical instability is due to an overlap of the low-spin Co3+/4+:t2g band with the top of the O2–:2p band, resulting in a removal of electron density from the O2–:2p band (i.e., oxidation of O2– ions) for (1 – x) < 0.5 in Li1–xCoO2 (Figure 3). (15, 16) In contrast, Mn offers excellent chemical stability as the high-spin Mn3+/4+:eg band lies well above the top of the O2–:2p band, Interestingly, Ni is between Mn and Co in structural and chemical stabilities as Ni3+ has higher OSSE than Mn3+ and the low-spin Ni3+/4+:eg band barely touches the top of the O2–:2p band. Furthermore, Co3+/4+:t2g6–x with a direct Co–Co interaction along the shared octahedral edges and a partially filled t2g band makes Li1–xCoO2 a metallic conductor for x > 0.1. In contrast, both Li1–xNiO2 and Li1–xMnO2 remain semiconductors for 0 ≤ (1 – x) ≤ 1.0 as the redox-active or partially filled eg band is involved in a 90° M–O–M (M = Mn or Ni) bonding. Nevertheless, with a high degree of Ni–O covalence, Li1–xNiO2 offers adequate electronic conductivity. With a 2-dimensional lithium ion diffusion, all three Li1–xMO2 (M = Mn, Co, and Ni) systems offer good lithium ion conduction. Also, Mn is the least expensive and least toxic while Co is the most expensive and most toxic among the three; Ni is in between. Considering the advantages and disadvantages among the three, the industry largely uses compositions, such as LiNi1/3Mn1/3Co1/3O2 (NMC-333), to realize the best possible among the three metal ions.
The LiMn2O4 spinel cathode with a three-dimensional structure and lithium ion diffusion offers high rate capability and good structural stability without phase transformations. It suffers, however, from a limited practical capacity (<120 A h kg–1) and manganese dissolution caused by a disproportionation of Mn3+ ions into Mn4+ and Mn2+ ions that is initiated by trace amounts of protons generated by a reaction of the lithium salt LiPF6 used in the electrolyte with trace amounts (ppm levels) of water present in the electrolyte. The olivine LiFeO4 cathode, on the other hand, offers good thermal stability and safety without oxygen release as the covalently bonded PO4 groups tightly hold the oxygen, but suffers from limited practical capacity (<160 A h kg–1), particularly limited volumetric capacity, lower operating voltage of ∼3.4 V, and poor electronic and lithium ion conductivity. Although the Fe2+/3+ redox couple lies at a much higher energy than the M3+/4+ (M = Mn, Co, and Ni) couples, the inductive effect, first recognized by Manthiram and Goodenough in the 1980s with polyanion cathodes, (17) lowers the Fe2+/3+ energy and increases the operating voltage to ∼3.4 V. The limited electronic and ionic conductivity have to be overcome by reducing the particle size to nanosize and coating with conductive carbon, which further decrease the already low volumetric energy density. The volumetric energy density is influenced by the crystallographic density of the structures. The crystallographic density decreases in the order layered > spinel > olivine. Therefore, among the three insertion-compound cathodes currently in play, the layered oxides are the ones that can provide the highest energy density.
Where Is Lithium Ion Technology Headed?
Increasing the Cell Voltage
There is tremendous interest to increase the energy density of lithium ion batteries by increasing the operating voltage or the charge-storage capacity or both. The only option to increase the cell voltage is raising the operating voltage of the cathode as the present anode (graphite) operating voltage is already close to that of Li/Li+. The three cathode structures (layered, spinel, and olivine) offer compositions with operating voltages higher than the currently used voltages of ∼4.3 V vs Li/Li+, (18) but the cathode surface with operating voltages >4.3 V is not stable in contact with the organic solvents EC, DEC, DMC, etc. used in the electrolyte. Examples of potential candidates with higher operating voltages are the spinel LiMn1.5Ni0.5O4 (∼4.7 V), (19) olivine LiCoPO4 (∼4.8 V), (14) and layered LiNi1–y–zMnyCozO2 with operating voltages >4.3 V to reversibly extract/insert more lithium. (20) Although the cathode–electrolyte interface is presently not stable above ∼4.3 V as the cathode redox energy lies below the HOMO of the electrolyte, it could potentially be circumvented by forming an optimum SEI on the cathode surface and thereby raising it above the HOMO of the electrolyte (Figure 3) analogous to that currently achieved with the graphite anode in commercial cells. While much concerted effort over the years has perfected the graphite anode, efforts toward stabilizing the cathode SEI are scarce. In fact, the electrolyte additives and compositions currently employed in commercial cells are largely tailored to making the graphite anode operable. The challenge is that any efforts made to make the cathode–electrolyte interface operable at higher voltages through electrolyte composition and/or additives should be compatible with the graphite anode; in other words, the approaches should not make the graphite–electrolyte interface unstable or damage the current stability achieved with the graphite–electrolyte interface.
Intuitive search for new electrolytes that are compatible with both the anode and cathode interfaces is needed if we are to increase the operating voltage. Organic solvents with compatible lithium salts that can offer a wider electrochemical stability window and support a higher operating voltage need to be developed. Solid electrolytes that support a wider electrochemical stability window are being intensively pursued, but the huge charge-transfer resistance at the solid–solid interface between the electrolyte and electrode and the mechanical stability and cost-effective, large-scale manufacturability of solid electrolytes pose problems. (21) Some examples of solid electrolytes pursued are based on garnet, LISICON, NASICON, sulfide, and poly(ethylene oxide) (PEO). (21) Development of new liquid or solid electrolytes with desired characteristics will enable the utilization of the high-voltage (>4.3 V) cathodes mentioned above and could also offer better safety.
Increasing the Charge-Storage Capacity
In the absence of a practically viable solution at present to increase the cathode operating voltage, much attention is being paid toward increasing the charge-storage capacities of both the anode and cathode. In this endeavor, anodes and cathodes that undergo a conversion reaction with lithium rather than an insertion reaction have drawn much attention in recent years. While the capacity of insertion-reaction electrodes is limited by the number of crystallographic sites available for reversible insertion/extraction of lithium, the conversion-reaction electrodes do not have such limitations. They display up to an order of magnitude higher capacities (Figure 1).
Examples of conversion-reaction anodes are Si, Sn, Sb, Ge, P, etc., offering much higher capacities than graphite (Figure 1). (2) They have higher operating voltages than graphite, which would lower the cell voltage, but anodes like Si operate at only a slightly higher voltage than graphite. The major challenges with the conversion-reaction anodes are the huge volume changes (up to ∼400% depending on the anode and the lithium content compared to <10% for graphite) occurring during the charge–discharge process, (22) pulverization of the particles, continuous formation of SEI, and the consequent trapping of active lithium from the cathode in the anode SEI. (23) Many approaches have been pursued, such as reducing the particle size to nanosize or deliberately leaving space within the active material architecture, but none of them are successful yet to be practically viable. (24, 25) The above approaches drastically increase SEI formation and decrease the volumetric energy density. The particle milling caused by volume changes results in a continuous formation of new surfaces during the charge–discharge process that further aggravates the formation of SEI. The only progress seen so far is incorporating a few % of Si into graphite to increase the charge storage capacity marginally in practical cells. It is a challenge to employ pure alloy anodes in practical cells that can offer adequate cycle life. An alternative is to use lithium metal as an anode, but reversible plating and stripping of lithium metal over a large number of cycles, SEI formation, and volume changes pose daunting challenges.
Examples of conversion-reaction cathodes are sulfur (or Li2S) and oxygen (or Li2O2 or Li2O), offering much higher capacities than layered, spinel, and olivine cathodes (Figure 1). However, they are met with numerous challenges. (26) The oxygen-based cathodes suffer from clogging by insoluble products, catalytic decomposition of electrolytes, moisture from air, and poor cycle life, making their practical viability extremely difficult, if not impossible. The challenges with sulfur-based cathodes are much less compared to those with oxygen, and much progress has been made in recent years in increasing the active material content and loading, suppressing dissolved polysulfide migration between the cathode and anode, and reducing the electrolyte amount. (27, 28) However, the necessity of pairing a lithium metal anode with sulfur or oxygen cathode poses formidable challenges, unless Li2S and Li2O2 cathodes could be successfully paired with an anode like graphite or Si or practical lithium-containing anodes that could be paired with sulfur or oxygen could be developed.
Recognizing the daunting challenges associated with the conversion-reaction electrodes, the recent focus has also been on near-term future technologies, i.e., toward increasing the capacity of insertion-reaction cathodes. In this regard, lithium-rich layered Li1+x(Ni1–y–zMnyCoz)1–xO2 oxides became appealing 15 years ago as they offer higher capacities of 250–300 A h kg–1. (29, 30) Unlike the conventional layered LiMO2 oxides, the lithium-rich layered oxides involve an oxidation first of the transition-metal ions to the 4+ state followed by an oxidation of oxide ions and an evolution of oxygen from the lattice during first charge. The potential participation of oxygen in the reversible redox process of Li1+x(Ni1–y–zMnyCoz)1–xO2 as well as in other lithium-rich materials, such as Li2Ru1–xSnxO2, Li1.211Mo0.467Co0.3O2, and Li2IrO3, have created much excitement and debate recently. (31-34) Unfortunately, despite intensive efforts for more than a decade, the lithium-rich Li1+x(Ni1–y–zMnyCoz)1–xO2 oxides suffer from layered to spinel phase transitions that are accompanied by a continuous voltage decay during cycling, inadequate cycle life, and inferior rate capability due to the presence of a significant amount of more localized Mn4+. Overall, the larger the discharge capacity and the amount of lithium extracted, the greater the tendency for Mn migration from the transition-metal layer to the lithium layer and voltage fade with cycling. (35) Although the potential of other lithium-rich oxides mentioned above and the practical viability of oxygen redox need to be fully assessed, it may prove challenging to realize the long cycle life needed, particularly for electric vehicles and grid storage, with significant amounts of holes in the O2–:2p band, i.e., formation of highly reactive peroxide or superoxide species could cause electrolyte oxidation and degrade cycle life; only time will clarify this predicament.
Focusing on High-Nickel Layered Oxides
With the challenges encountered with lithium-rich Li1+x(Ni1–y–zMnyCoz)1–xO2 cathodes, much attention is currently being directed toward increasing the capacity by increasing the Ni content in layered LiNi1–y–zMnyCozO2. The high-nickel cathodes are emerging as a near-term future technology. As discussed in the previous section, the characteristics of Ni are between those of Co and Mn in almost all the necessary aspects (chemical stability, structural stability, conductivity, cost, and toxicity). More importantly, Ni3+ can be fully oxidized to Ni4+ without the loss of oxygen from the lattice, unlike in the case of Co3+. (15, 16) Therefore, LiNiO2 is a better preferred layered oxide cathode. Unfortunately, LiNiO2 encounters a different set of challenges. First, it is very difficult to keep all Ni as Ni3+ during the synthesis process at higher temperatures (>700 °C), so the existence of part of Ni as Ni2+ results in a volatilization of part of lithium and formation of a lithium-deficient Li1–xNi1+xO2. This implies a cation disorder between Li and Ni and the presence of Ni in the lithium layer can impede the rate capability. Second, LiNiO2 undergoes a series of phase transitions during the charge–discharge process, particularly at deep charge involving the removal of a significant amount of lithium from the lattice. This, again, can lead to a degradation in rate capability. Third, Ni4+ is highly oxidizing and reacts aggressively with the organic electrolytes used in lithium ion cells. The reaction results in the formation of a thick SEI layer, which, again, degrades the rate capability, increases the impedance, and consumes active lithium. Fourth, the chemical instability of the highly oxidized Ni4+ results in a transformation of the layered oxide to a rock salt LixNi1–xO phase on the surface of LiNiO2. Fifth, the highly oxidized and unstable Ni4+ also causes concern with thermal runaway. Because of these challenges, LiNiO2 was largely ignored as a possible cathode for decades. The push to increase the energy density, the potential to obtain higher capacity as Ni3+ could be oxidized all the way to Ni4+, and the unsolvable problem of the voltage decay associated with the lithium-rich layered oxides have reinvigorated the interest in high-nickel-content oxides during the past couple of years.
With the renewed interest in LiNiO2, the industry has been slowly moving from LiNi1/3Mn1/3Co1/3O2 to increase the Ni content in LiNi1–y–zMnyCozO2. For example, compositions such as LiNi0.4Mn0.3Co0.3O2 (NMC-433) and LiNi0.6Mn0.2Co0.2O2 (NMC-622) have become or are becoming commercial now. The driving force to successively increase the Ni content is the ability to increase the capacity, tap density, and volumetric energy density; with Ni contents of ∼0.9, practical capacities as high as ∼230 A h kg–1 could be realized. However, the long-term stability of NMC-622 for thousands of cycles for applications such as electric vehicles still needs to be established. The problems become increasingly serious as the Ni content increases further beyond NMC-622 to NMC-811 or higher. The high-nickel LiNi0.8Co0.15Al0.05O2 (NCA) is used in commercial cells, but the high Al content decreases the practical capacity to ∼180 A h kg–1, and NCA suffers from gas evolution issues during cycling.
During the past couple of years, significant understanding has been made on high-nickel cathodes with advanced analytical techniques, which is extremely valuable if we were to successfully employ nickel-rich layered oxides as we move forward to increase the energy density. An in-depth characterization of LiNi0.7Mn0.15Co0.15O2 (NMC-71515), before and after cycling in 1 M LiPF6 in EC-DEC electrolyte, with a combination of X-ray photoelectron spectroscopy (XPS), time-of-flight secondary ion mass spectroscopy (TOF-SIMS), and high-resolution transmission electron microscopy (HR-TEM) reveals that the SEI layer from the cathode surface to the exterior is successively composed of the rock salt LixNi1–xO phase, transition-metal fluorides formed by dissolved metal ions, and organic liquid electrolyte decomposition products. (36) Also, the SEI layer grows continuously with cycling. Figure 4a illustrates a TOF-SIMS chemical mapping of the organic electrolyte decomposition products (7Li2+/7Li2F and 7LiF2–) and transition-metal fluorides (MnF3–) on a secondary particle of NMC-71515 cathode after cycling. Figure 4b shows a TOF-SIMS comparison of the dissolved transition-metal ions (MnF3–) on two high-Ni cathodes, LiNi0.61Mn0.27Co0.12O2 (undoped NMC with no Al) and LiNi0.60Mn0.27Co0.12Al0.01O2 (1 mol % Al-doped NMC) before and after 3,000 cycles in a full cell with graphite anode and 1.2 M LiPF6 in EC-EMC with 1 wt % vinylene carbonate (VC) electrolyte. (37) It is remarkable that doping with 1 mol % Al drastically suppresses metal ion dissolution from the cathode as the covalent Al–O bonds keep the oxygen tightly bonded, lower the basicity, and discourage transition-metal ion dissolution by acidic attack. Figure 4c compares the amounts of dissolved transition-metal ions and lithium dendrite on the two graphite anodes, one paired with undoped NMC with no Al and the other paired with 1 mol % Al-doped NMC, after 3,000 cycles. (37) It is amazing that the cell with 1 mol % Al-doped cathode has drastically reduced dissolved metal ions and lithium metal plating/dendrite on the graphite anode. Figure 4d schematically shows the buildup of the SEI and plated Li on the graphite anode. The drastic reduction in trapped active lithium in the form of dendrite, enabled by a suppressed metal ion dissolution, leads to superior cycle life over 3,000 cycles for the cell with 1 mol % Al-doped NMC cathode compared to that with the undoped NMC cathode. (37)
Figure 4
Figure 4. (a) TOF-SIMS chemical mapping of the organic electrolyte decomposition layer and dissolved transition-metal layer in the form of fluorides on an NMC cathode particle. (b) Comparison after 3,000 cycles of the amounts of transition-metal dissolution, forming metal fluorides (e.g., MnF2), from an undoped and a 1 mol % Al-doped NMC cathode relative to that from a fresh electrode. (c) Comparison after 3,000 cycles of the amounts of dissolved transition metals and the calculated thickness of Li metal dendrites on graphite anodes that were paired with an undoped and a 1 mol % Al-doped NMC cathode. (d) Schematic illustrating the evolution of the SEI on graphite anode during cycling under the influence of dissolved transition-metal ion crossover from the cathode to the anode. Reproduced with permission from ref 37. Copyright 2017 American Chemical Society.
Significant performance gains are being realized with stabilized high-nickel layered oxide cathodes through compositional control, including doping and concentration-gradient structures with less Ni on the surface. (37, 38) The salt and solvents in the electrolyte also play a dominant role on cathode surface reactivity, SEI formation, metal ion dissolution, cycle life, rate capability, and energy density. Optimal electrolyte compositions that are compatible with and support favorable SEI formation on both the cathode and anode not only could enhance the cycle life under the current operating conditions of <4.3 V but could also enable operation to higher voltages of layered oxide cathodes as well as other cathodes like spinel LiMn1.5Ni0.5O4 and olivine LiCoPO4.
Conclusions
The current lithium ion technology based on insertion-reaction cathodes and anodes will continue for the foreseeable future, despite their limited energy density dictated by the number of crystallographic sites available as well as the structural and chemical instabilities at deep charge. Much effort has been made toward conversion-reaction anodes and cathodes as they offer up to an order of magnitude higher capacities than insertion-reaction electrodes, but their practical viability is met with challenges. Renewed interest in employing lithium metal as an anode and replacing liquid electrolytes with a solid electrolyte has emerged recently as they can offer safer cells with higher operating voltages and charge-storage capacity, but only time will reveal their practical viability. With the challenges encountered with the alternatives (conversion-reaction electrodes, lithium metal, and solid electrolytes), a feasible near-term strategy is to focus on high-nickel layered oxide cathodes, liquid electrolytes compatible with and forming stable SEI on both graphite anode and high-Ni cathodes, innovations in cell engineering to fabricate thicker electrodes and reduce inactive components, and novel system integration to realize safer, long-life, affordable systems.